论文分享
【国内论文】南京大学——掺杂铟的 β-Ga₂O₃ 电子迁移率增强的第一性原理研究
日期:2025-03-06阅读:967
由南京大学的研究团队在学术期刊 Physical Chemistry Chemical Physics 发布了一篇名为 First principles investigation of electron mobility enhancement of β-Ga2O3 doped with indium(掺杂铟的 β-Ga2O3 电子迁移率增强的第一性原理研究)的文章。
摘要
β-Ga2O3 是近年来备受关注的新一代宽禁带半导体材料之一。然而,其在室温下的电子迁移率明显低于 GaN 和 SiC。通过合金化 β-Ga2O3 预计能够赋予材料更优异的载流子传输特性。因此,本研究基于第一性原理,考虑声学形变势(ADP)散射、极化光学声子(POP)散射和离化杂质(IMP)散射,对纯 Ga2O3、In 掺杂 Ga2O3 和 Al 掺杂 Ga2O3 的电子迁移率进行了深入研究。对材料的结构优化、电子能带结构以及温度和载流子浓度对电子迁移率的影响进行了系统分析。结果表明,在 105–650 K 温度范围内,In–Ga2O3 的电子迁移率始终最高,且在 150–650 K 之间,POP 散射是限制电子迁移率的主要因素。In 掺杂提升电子迁移率的原因主要归因于 In 5s 电子态引起的较小有效质量,尽管其电子-声子耦合强度略有增加。在 1.0 × 1017 cm−3 电子浓度下,β-Ga2O3、Al–Ga2O3 和 In–Ga2O3 在室温(300 K)下的预测电子迁移率分别为 151.5 cm2 V−1 s−1、137.8 cm2 V−1 s−1 和 184.9 cm2 V−1 s−1。该研究为提高 β-Ga2O3 电子迁移率提供了一种可行的方案,并为工程化调控其电子传输特性以应用于高功率电子器件提供了指导。
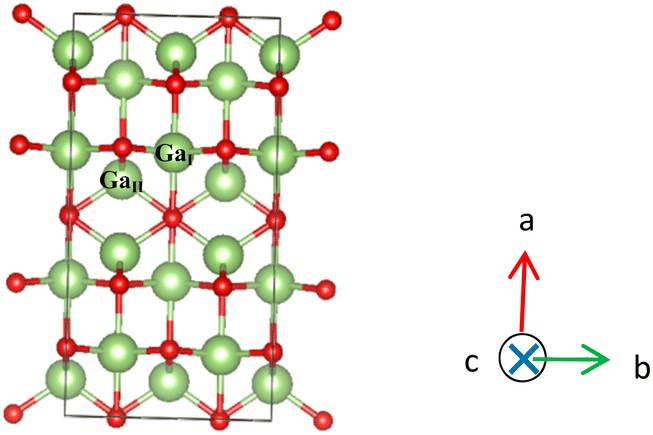
图 1. Ga2O3 计算模型的晶体结构。绿色球体代表阳离子锑,红色球体代表阴离子氧。
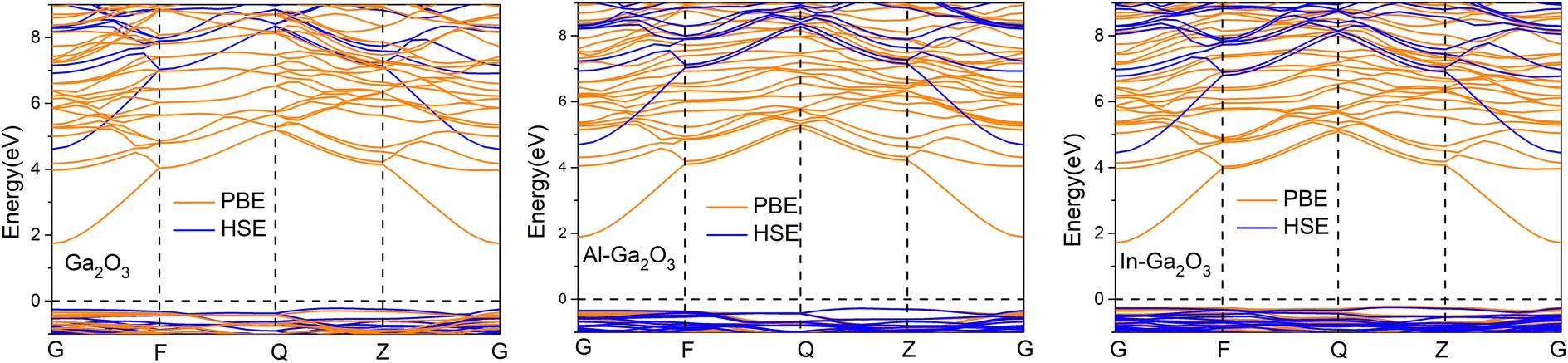
图 2 使用 PBE(橙线)和 HSE(蓝线)函数计算的纯 Ga2O3、Al-Ga2O3 和 In-Ga2O3 的电子能带结构。
DOI:
doi.org/10.1039/D4CP04220D