Paper Sharing
【Domestic Papers】Theoretical investigation of electronic structure and thermoelectric properties of AlGa point defects in two-dimensional β-Ga₂O₃
日期:2025-10-20阅读:543
Researchers from the Shihezi University have published a dissertation titled "Theoretical investigation of electronic structure and thermoelectric properties of AlGa point defects in two-dimensional β-Ga2O3" in Results in Physics.
Project Support
This work was financially supported by National Natural Science Foundation of China (Grant nos. 62104162, 12264041, 12164038).
Background
Thermoelectric materials, which enable direct interconversion between thermal and electrical energy, hold unique value in waste heat recovery and solid-state refrigeration technologies. Among emerging wide-bandgap semiconductors, two-dimensional (2D) monoclinic gallium oxide (β-Ga2O3) has attracted significant attention due to its ultra-low lattice thermal conductivity (0.71 W/(m·K) at 300 K, nearly 20 times lower than bulk β-Ga2O3). H. J. Wu et al. reported a sucrose-derived β-Ga2O3 with intentionally engineered porosity through low-temperature sintering (600 °C), achieving low thermal conductivity of 0.45 W/(m·K) (room temperature), which further decreases to 0.29 W/(m·K) at 600 °C. In addition, the intrinsic wide bandgap of β-Ga2O3 in bulk crystals and thin films, which is typically 4.70 eV ∼ 4.85 eV at room temperature, fundamentally limits carrier concentration and electrical conductivity. Studies show 2D β-Ga2O3 is an indirect-bandgap semiconductor like bulk β-Ga2O3. The bandgaps of 2D β-Ga2O3 calculated by GGA, HSE and GGA + U methods are 2.30 eV,4.69 eV and 4.66 eV respectively. Nan Chen et al. calculated the band gap of 2D β-Ga2O3 to be 1.89 eV using the PBE method, and 4.91 eV using the GGA + U method. The researchers found that defects can regulate the bandgap of β-Ga2O3.
Abstract
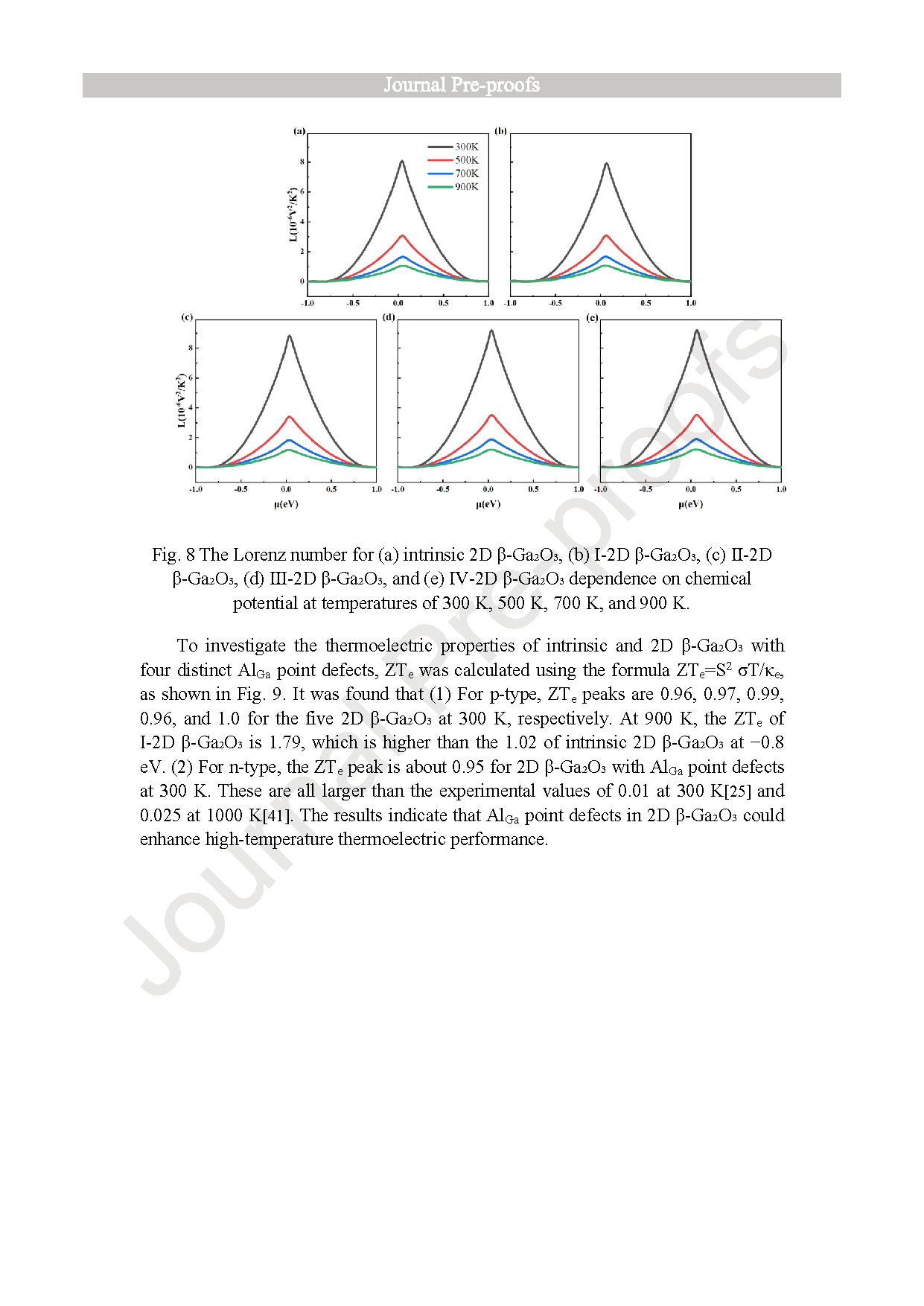
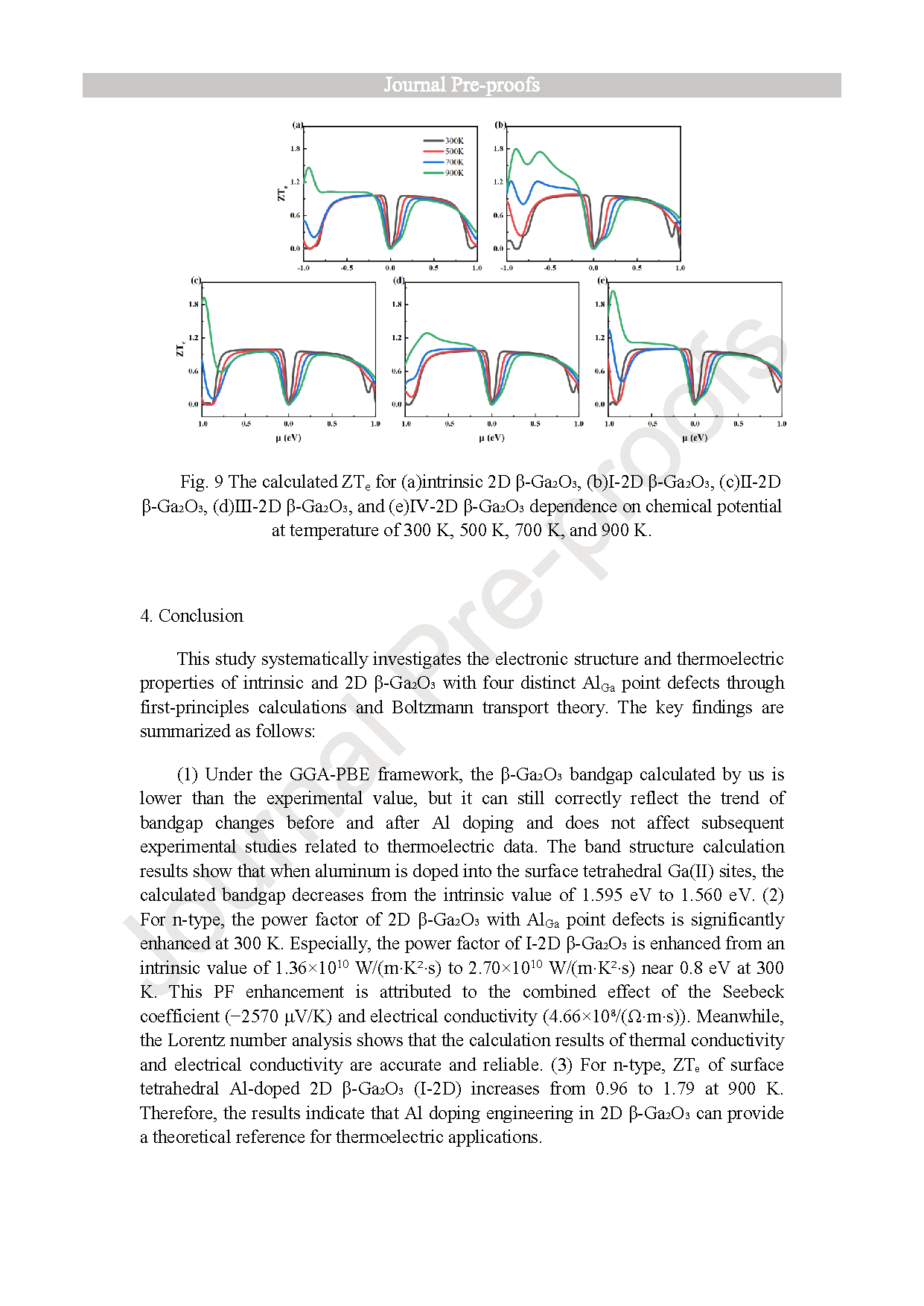
As a wide-bandgap semiconductor, β-Ga2O3 possesses excellent physical and chemical properties, such as ultra-low lattice thermal conductivity for thermoelectric applications. We investigated the electronic structure and thermoelectric properties of two-dimensional (2D) β-Ga2O3 with Al substitutions at four sites (surface/inner tetrahedral Ga(I) and octahedral Ga(II)) using density-functional theory (DFT) and Boltzmann transport theory. It was found that: (1) Compared to intrinsic 2D β-Ga2O3, the bandgap of surface tetrahedral Al-doped 2D β-Ga2O3 (I-2D) is reduced by 0.035 eV, and the density of states (DOS) for four distinct AlGa point defects in 2D β-Ga2O3 indicates that Al(p) orbitals contribute mainly to the valence band maximum (VBM). (2) 2D β-Ga2O3 with AlGa enhances n-type Seebeck coefficients. Especially, I-2D achieves the maximum power factor of 2.70 × 1010 W/(m·K2·s) of power factor for n-type near 0.8 eV (300 K), which is higher than the intrinsic value of 1.36 × 1010 W/(m·K2·s). (3) For n-type, Ⅰ-2D β-Ga2O3, the electrical conductivity is 4.66 × 1018/(Ω·m·s), at 1.0 eV and the electronic thermal conductivity (κe) is 1.9 × 1013 W/(m·K) near 0.8 eV at 300 K. The Lorenz numbers for the five 2D β-Ga2O3 structures all conform to the Lorenz distribution, indicating that the calculation results of thermal conductivity and electrical conductivity are accurate and reliable. (4) Type I-2D β-Ga2O3 achieves a peak n-type ZTe of 1.79 at 900 K (vs. 0.96 for intrinsic). The results demonstrate that Al doping in 2D β-Ga2O3 optimizes thermoelectric properties for applications.
Highlights
● Electronic structure regulation: In Al-doped 2D β-Ga2O3, surface tetrahedral doping (Ⅰ-2D) reduces the bandgap by 0.035 eV. Al(p) orbitals contribute significantly to the valence band maximum (VBM). All Al-doped structures exhibit negative formation energies, indicating the thermodynamic spontaneity of the doping process.
● Optimization of thermoelectric properties: Al doping significantly enhances the n-type Seebeck coefficient. Specifically, Ⅰ-2D achieves a power factor of 2.70 × 1010 W/(mK2s) near 0.8 eV at 300 K, which is twice the intrinsic value. Its n-type electrical conductivity reaches 4.66 × 1018/(Ωms) at 1.0 eV, and the electronic thermal conductivity (κe) is 1.9 × 1013 W/(mK) near 0.8 eV.
● Significant improvement in ZTe: Ⅰ-2D attains a peak n-type ZTe of 1.79 at 900 K, much higher than the intrinsic structure’s value of 0.96. The Lorenz numbers conform to the theoretical distribution, verifying the reliability of the calculated electrical and thermal conductivities, suggesting that Al-doped 2D β-Ga2O3 is a promising material for high-efficiency thermoelectric applications.
Conclusion
This study systematically investigates the electronic structure and thermoelectric properties of intrinsic and 2D β-Ga2O3 with four distinct AlGa point defects through first-principles calculations and Boltzmann transport theory. The key findings are summarized as follows:
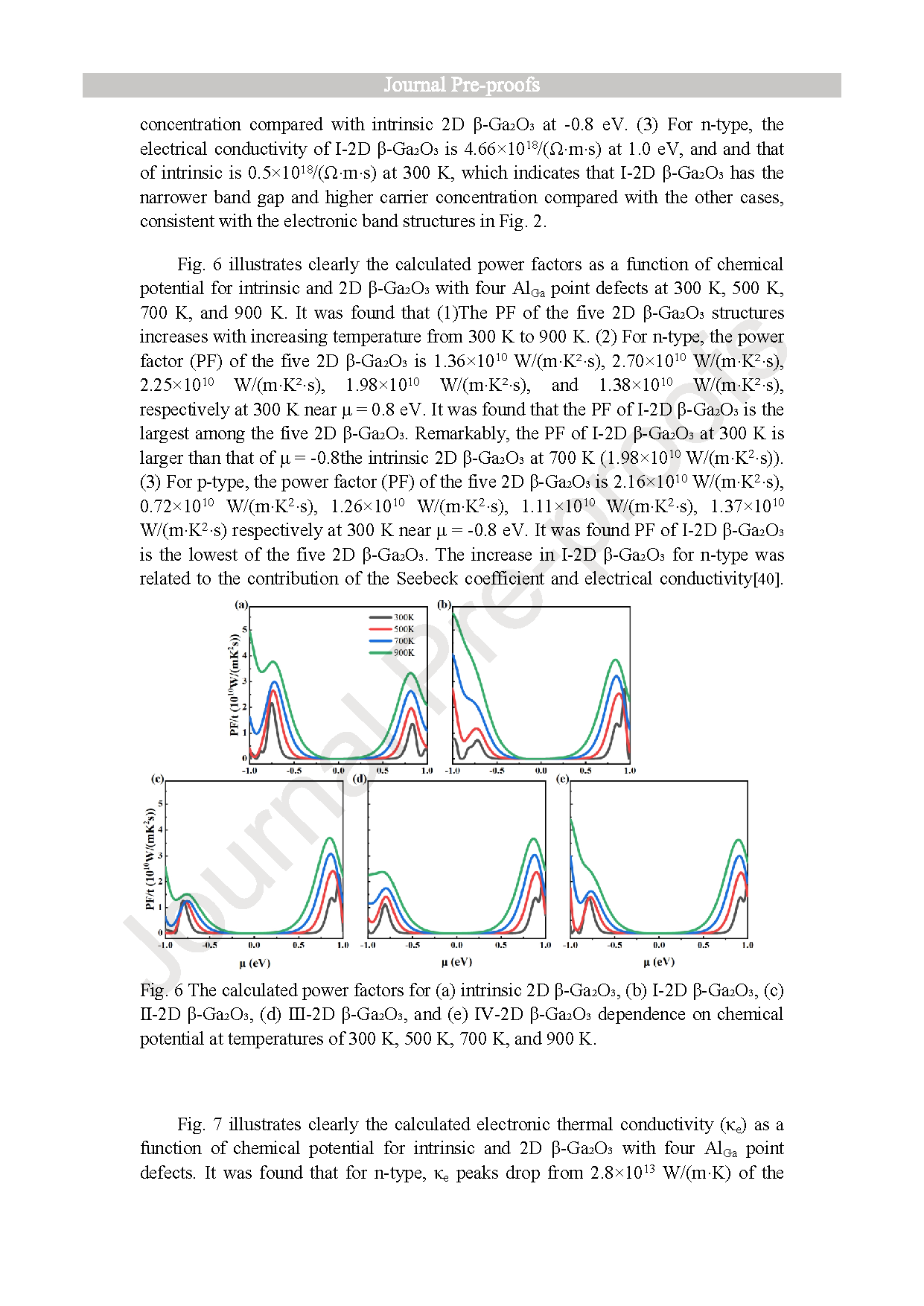
(1) Under the GGA-PBE framework, the β-Ga2O3 bandgap calculated by us is lower than the experimental value, but it can still correctly reflect the trend of bandgap changes before and after Al doping and does not affect subsequent experimental studies related to thermoelectric data. The band structure calculation results show that when aluminum is doped into the surface tetrahedral Ga(II) sites, the calculated bandgap decreases from the intrinsic value of 1.595 eV to 1.560 eV. (2) For n-type, the power factor of 2D β-Ga2O3 with AlGa point defects is significantly enhanced at 300 K. Especially, the power factor of Ⅰ-2D β-Ga2O3 is enhanced from an intrinsic value of 1.36 × 1010 W/(m·K2·s) to 2.70 × 1010 W/(m·K2·s) near 0.8 eV at 300 K. This PF enhancement is attributed to the combined effect of the Seebeck coefficient (−2570 μV/K) and electrical conductivity (4.66 × 108/(Ω·m·s)). Meanwhile, the Lorentz number analysis shows that the calculation results of thermal conductivity and electrical conductivity are accurate and reliable. (3) For n-type, ZTe of surface tetrahedral Al-doped 2D β-Ga2O3 (Ⅰ-2D) increases from 0.96 to 1.79 at 900 K. Therefore, the results indicate that Al doping engineering in 2D β-Ga2O3 can provide a theoretical reference for thermoelectric applications.
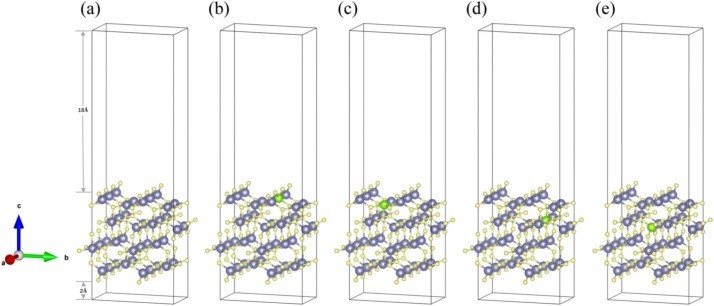
Fig. 1. Structural models of (a)intrinsic 2D β-Ga2O3, (b)Ⅰ-2D β-Ga2O3, (c)Ⅱ-2D β-Ga2O3, (d)Ⅲ-2D β-Ga2O3, and (e)Ⅳ-2D β-Ga2O3.
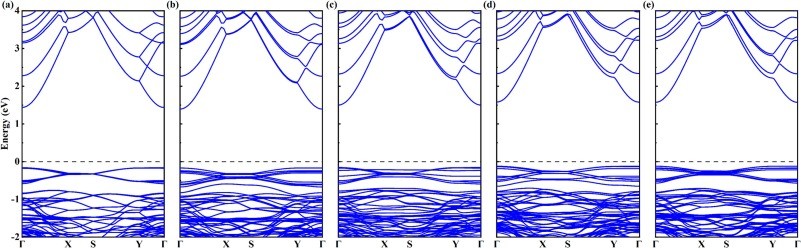
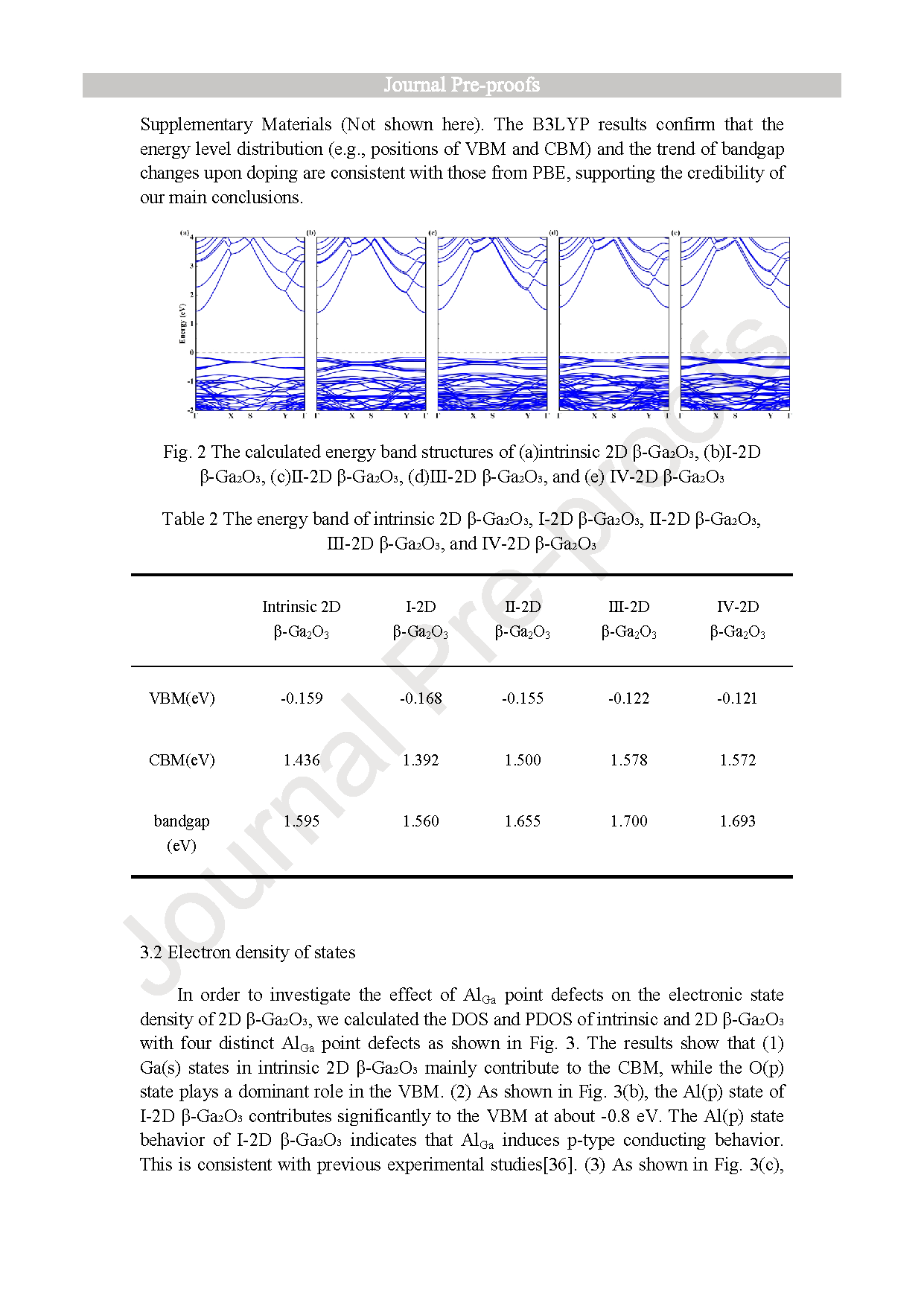
Fig. 2. The calculated energy band structures of (a)intrinsic 2D β-Ga2O3, (b)Ⅰ-2D β-Ga2O3, (c)Ⅱ-2D β-Ga2O3, (d)Ⅲ-2D β-Ga2O3, and (e) Ⅳ-2D β-Ga2O3.
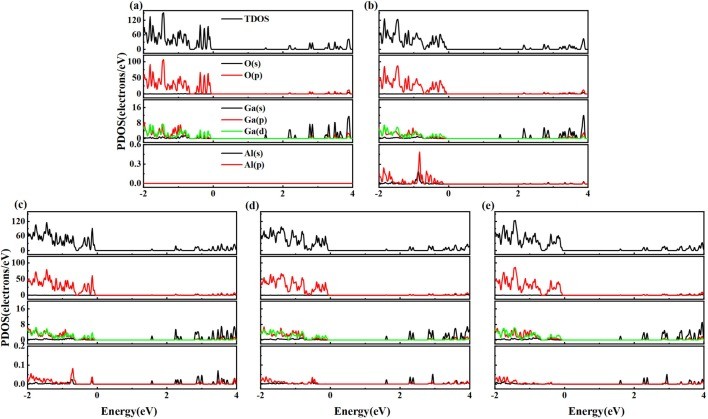
Fig. 3. The total density of states (TDOS) and partial density of states (PDOS) for (a)intrinsic 2D β-Ga2O3, (b)Ⅰ-2D β-Ga2O3, (c)Ⅱ-2D β-Ga2O3, (d)Ⅲ-2D β-Ga2O3, and (e)Ⅳ-2D β-Ga2O3.
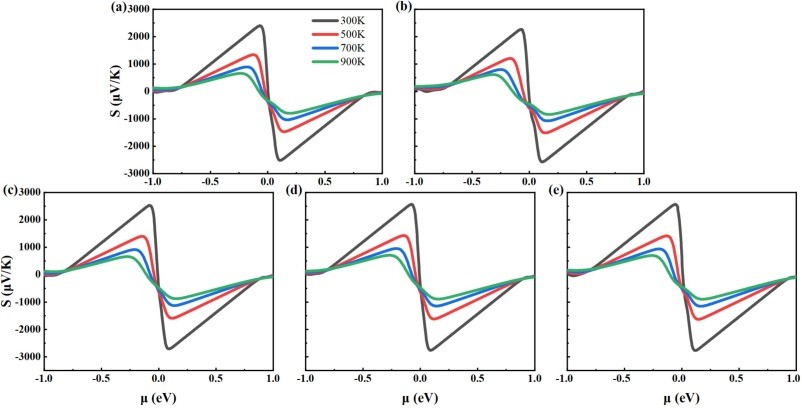
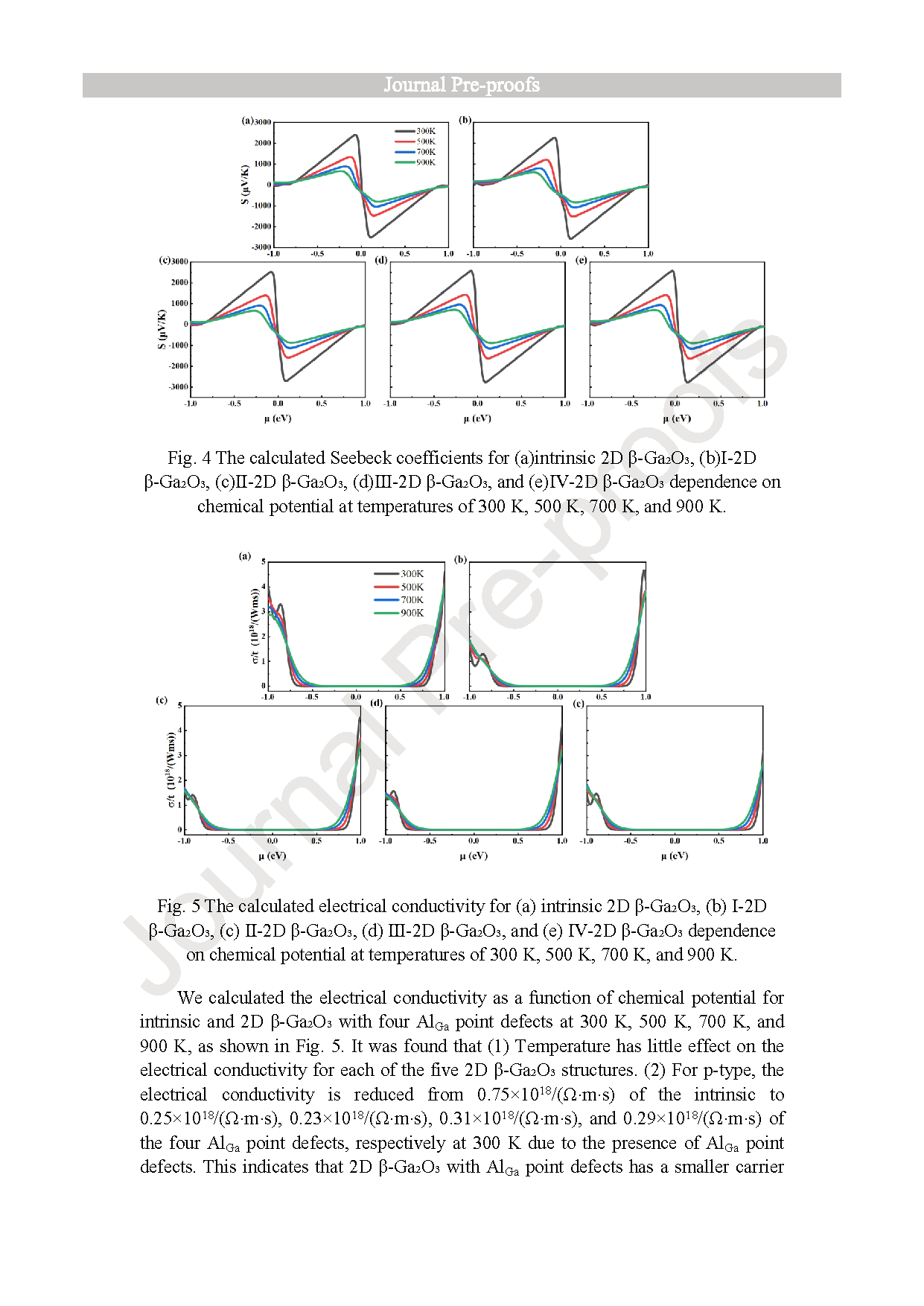
Fig. 4. The calculated Seebeck coefficients for (a)intrinsic 2D β-Ga2O3, (b)Ⅰ-2D β-Ga2O3, (c)Ⅱ-2D β-Ga2O3, (d)Ⅲ-2D β-Ga2O3, and (e)Ⅳ-2D β-Ga2O3 dependence on chemical potential at temperatures of 300 K, 500 K, 700 K, and 900 K.
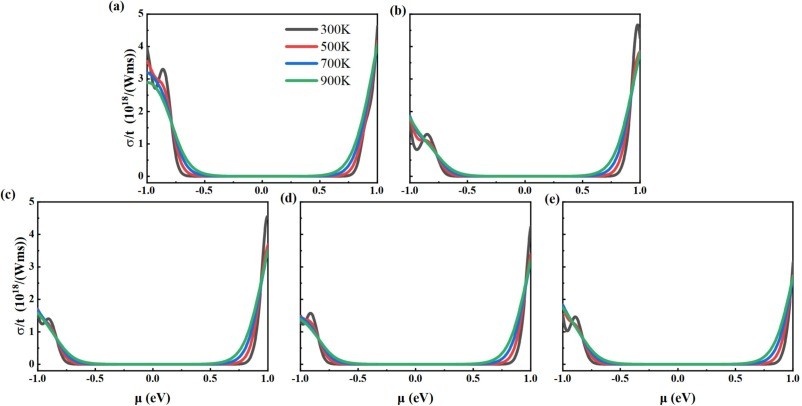
Fig. 5. The calculated electrical conductivity for (a) intrinsic 2D β-Ga2O3, (b) Ⅰ-2D β-Ga2O3, (c) Ⅱ-2D β-Ga2O3, (d) Ⅲ-2D β-Ga2O3, and (e) Ⅳ-2D β-Ga2O3 dependence on chemical potential at temperatures of 300 K, 500 K, 700 K, and 900 K.
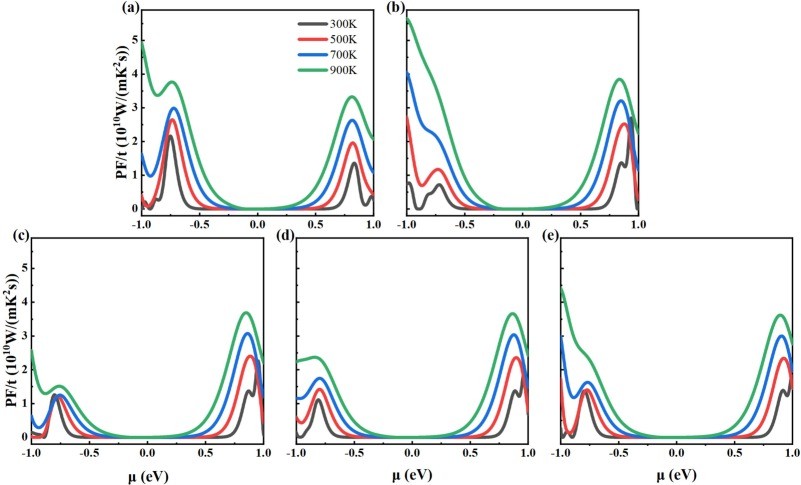
Fig. 6. The calculated power factors for (a) intrinsic 2D β-Ga2O3, (b) Ⅰ-2D β-Ga2O3, (c) Ⅱ-2D β-Ga2O3, (d) Ⅲ-2D β-Ga2O3, and (e) Ⅳ-2D β-Ga2O3 dependence on chemical potential at temperatures of 300 K, 500 K, 700 K, and 900 K.
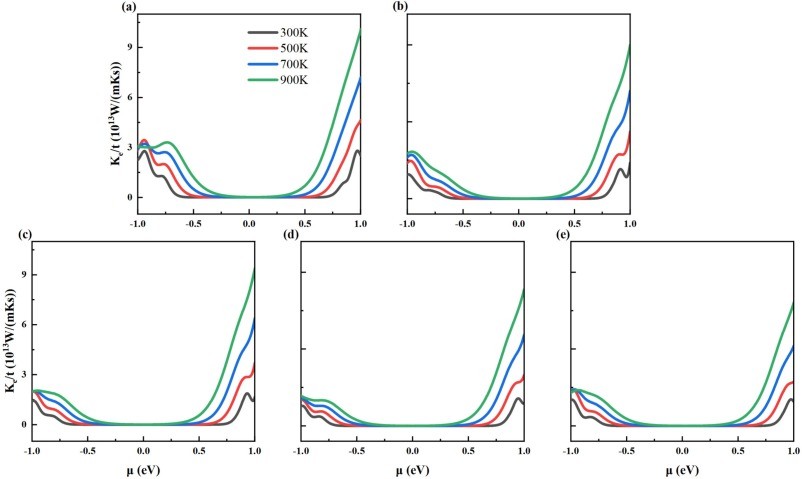
Fig. 7. The calculated electronic thermal conductivity for (a) intrinsic 2D β-Ga2O3, (b) Ⅰ-2D β-Ga2O3, (c) Ⅱ-2D β-Ga2O3, (d) Ⅲ-2D β-Ga2O3, and (e) Ⅳ-2D β-Ga2O3 dependence on chemical potential at temperature of 300 K, 500 K, 700 K, and 900 K.
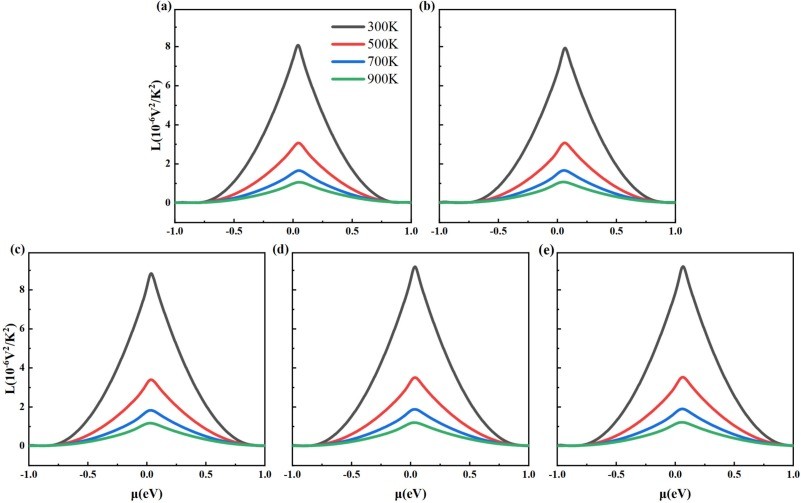
Fig. 8. The Lorenz number for (a) intrinsic 2D β-Ga2O3, (b) Ⅰ-2D β-Ga2O3, (c) Ⅱ-2D β-Ga2O3, (d) Ⅲ-2D β-Ga2O3, and (e) Ⅳ-2D β-Ga2O3 dependence on chemical potential at temperatures of 300 K, 500 K, 700 K, and 900 K.
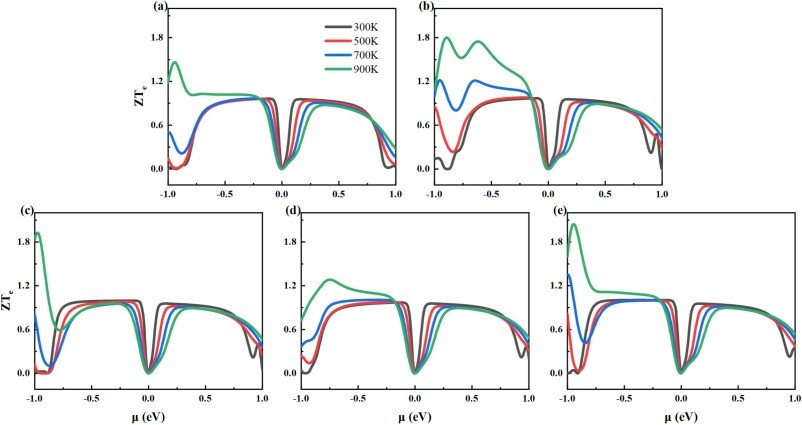
Fig. 9. The calculated ZTe for (a)intrinsic 2D β-Ga2O3, (b)Ⅰ-2D β-Ga2O3, (c)Ⅱ-2D β-Ga2O3, (d)Ⅲ-2D β-Ga2O3, and (e)Ⅳ-2D β-Ga2O3 dependence on chemical potential at temperature of 300 K, 500 K, 700 K, and 900 K.
DOI:
doi.org/10.1016/j.rinp.2025.108424