Paper Sharing
【Domestic Papers】Dependence of Interfacial Bonding Strength on Crystallographic Matching in β-Ga₂O₃/Diamond Heterogeneous Bonding
日期:2026-03-23阅读:283
Researchers from the Jiangsu Provincial Key Laboratory of Food Advanced Manufacturing Equipment Technology, School of Mechanical Engineering, Jiangnan University have published a dissertation titled "Dependence of Interfacial Bonding Strength on Crystallographic Matching in β-Ga₂O₃/Diamond Heterogeneous Bonding" in Acta Physica Sinica.
Background
β-Ga₂O₃ is a representative fourth-generation ultra-wide-bandgap semiconductor with a bandgap of 4.6–4.9 eV and a breakdown electric field of up to 8 MV/cm, showing great potential for next-generation power switching devices. It can be fabricated into high-quality, large-size and low-cost substrates via melt growth technology. However, its intrinsic thermal conductivity is only 10–27 W/(m·K), and the severe self-heating effect restricts the performance and reliability of devices. Heterogeneous bonding of β-Ga₂O₃ with diamond (thermal conductivity > 2000 W/(m·K)) is a key solution to break through the heat dissipation bottleneck. Existing experiments have achieved atomically flat bonding between the two materials, but the atomic-scale mechanism of interfacial microevolution and crystal orientation dependence of bonding strength remains unclear. Traditional density functional theory (DFT) has high accuracy but high computational cost, making it difficult to simulate large-atom interfaces; molecular dynamics (MD) can handle large-scale systems, but traditional empirical potentials lack accuracy in describing complex oxides and covalent bond recombination. Machine learning interatomic potentials (MLIPs) balance accuracy and efficiency, among which the moment tensor potential (MTP) has significant advantages in simulating complex oxide interfaces. The team carried out research to reveal the interfacial bonding rules based on this.
Abstract
As a representative fourth-generation ultra-wide-bandgap semiconductor, β-Ga₂O₃ has shown great promise for high-voltage, high-frequency, and high-power electronic devices because of its ultra-wide bandgap, high breakdown electric field, and low fabrication cost. However, its intrinsically low thermal conductivity makes it difficult to dissipate heat efficiently during device operation. This severely limits its performance and reliability under high power density. In recent years, heterogeneous bonding of β-Ga₂O₃ with high-thermal-conductivity materials such as diamond has emerged as an important strategy to overcome this thermal bottleneck. Such structures can potentially combine excellent electrical properties with efficient thermal management. However, the microscopic evolution of the β-Ga₂O₃/diamond interface and the crystal-orientation dependence of interfacial bonding strength remain unclear. This knowledge gap has hindered interface design, bonding process optimization, and reliability improvement. To address these issues, this study combined large-scale molecular dynamics simulations with density functional theory calculations. A million-atom training dataset with first-principles accuracy was constructed. Based on this dataset, a high-accuracy Moment Tensor Potential (MTP) was developed. Using the fitted potential, we systematically investigated the interfacial structural evolution of β-Ga₂O₃/diamond heterogeneous bonding models containing an amorphous transition layer during relaxation. We also examined how interfacial bonding strength depends on different crystal orientation combinations. The results show that, for all orientation combinations, the amorphous interlayer effectively relieves lattice mismatch and local stress concentration during relaxation. As a result, atomically dense contact is achieved at the bonded interface without obvious defects such as voids, cracks, or delamination. These findings indicate that the introduction of an amorphous interlayer improves interfacial structural integrity and bonding stability. Further mechanical analysis shows that the β-Ga₂O₃ (010)/diamond (100) configuration exhibits the best interfacial bonding strength and overall mechanical performance, demonstrating a clear orientation-matching advantage. This work reveals, at the atomic scale, the evolution mechanism of β-Ga₂O₃/diamond heterogeneous bonding interfaces and the crystal-orientation dependence of their bonding strength. It provides important theoretical guidance for interface design, wafer bonding process optimization, and the improvement of mechanical stability and long-term reliability in high-power devices.
Highlights
Potential Development: Constructed a million-atom DFT-precision dataset for the C-O-Ga ternary system and developed an MTP machine learning potential suitable for β-Ga₂O₃/diamond heterogeneous bonding, balancing simulation accuracy and efficiency.
Interface Simulation: Constructed a heterogeneous bonding model with an amorphous transition layer, revealed the interfacial atomic reconstruction law at 500 K, and achieved atomically dense and defect-free bonding for all crystal orientation combinations.
Crystal Orientation Optimization: Quantitatively characterized the interfacial mechanical properties, determined that β-Ga₂O₃ (010)/diamond (100) is the optimal crystal orientation matching, with a tensile strength close to 5 GPa and the optimal thermal resistance.
Mechanism Revelation: Clarified the mechanism of the amorphous layer alleviating lattice mismatch and optimizing stress distribution and established the correlation between bonding strength and crystal orientation matching.
Conclusion
In this study, we constructed a million-atom training dataset with first-principles accuracy, and based on this dataset, used the MTP framework to develop a machine-learning interatomic potential suitable for the C-O-Ga ternary system. Using this high-performance potential, we systematically simulated and predicted the interfacial evolution process of β-Ga₂O₃ and diamond heterogeneous bonding under various crystal plane combinations. By quantitatively evaluating the interfacial bonding strength and maximum strain, the anisotropic characteristics of interfacial bonding performance were revealed, and the optimal mechanical crystal plane matching scheme was selected as the β-Ga₂O₃ (010)/diamond (100) combination. In addition, for the β-Ga₂O₃ (100) and β-Ga₂O₃ (001) crystal planes, the diamond (111) crystal plane is the optimal choice. The significance of this study is that a high-precision machine learning potential for β-Ga₂O₃/diamond heterogeneous bonding is constructed, which can accurately describe the complex chemical bonding process and mechanical properties at the interface, and provides a theoretical basis for further optimizing the heterogeneous integration process of β-Ga₂O₃ and diamond. In addition, the machine learning potential used in this study provides a reliable tool for efficient simulation of large-scale heterogeneous interface structures and can be widely extended to the prediction and design of interfacial behavior of other new semiconductor heterojunctions in the future, thus strongly promoting the in-depth integration of material science mechanism exploration and device engineering applications.
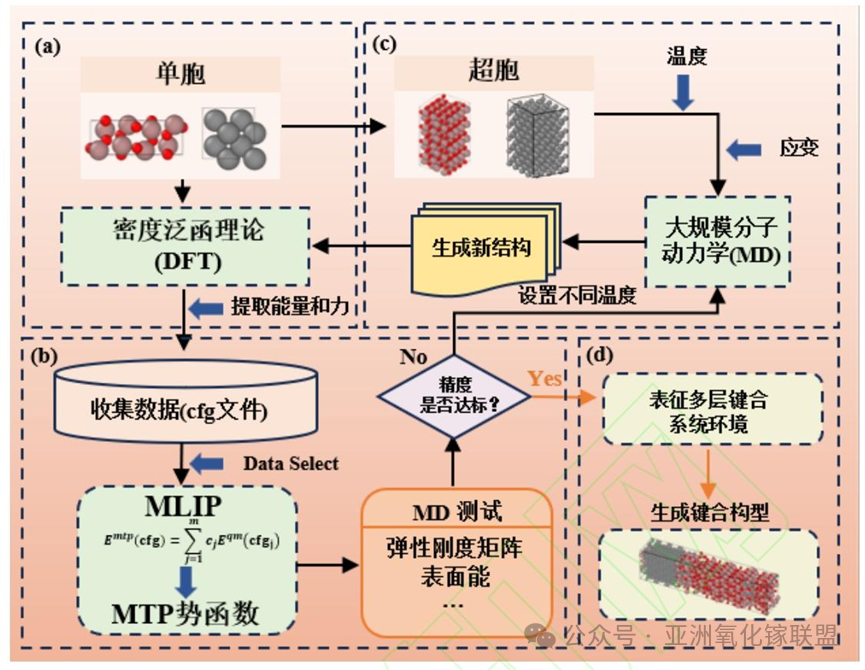
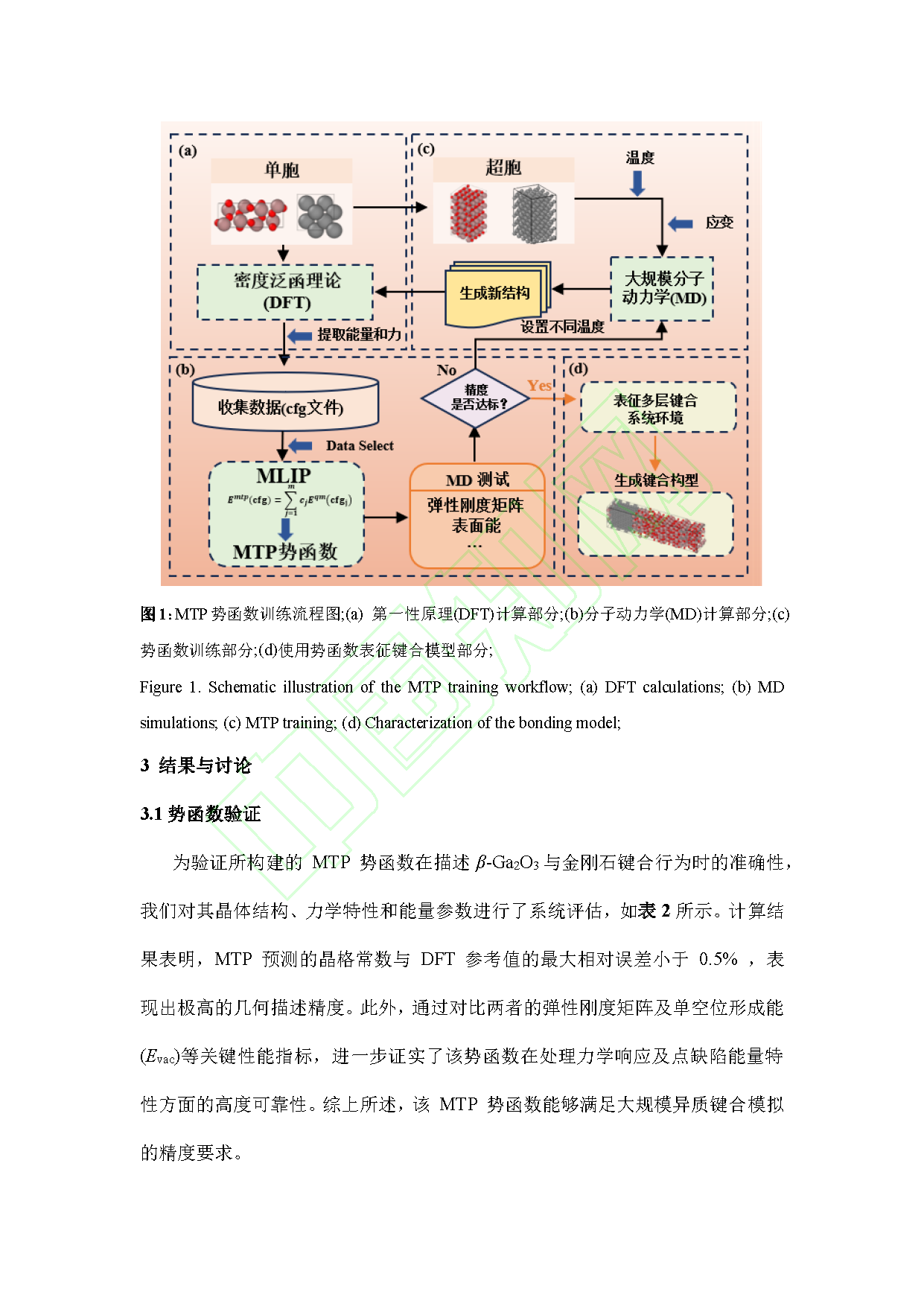
Fig. 1. Schematic illustration of the MTP training workflow; (a) DFT calculations; (b) MD simulations; (c) MTP training; (d) Characterization of the bonding model.
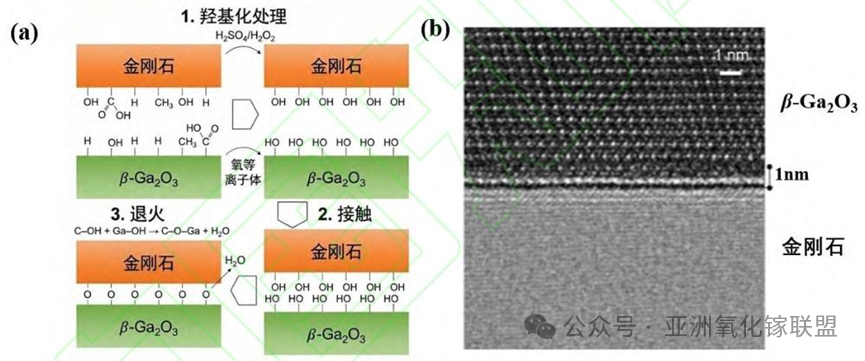
Fig. 2. Bonding mechanism and SEM characterization. (a) Hydrophilic bonding schematic at 500 K; (b) SEM image of the interface revealing a ~1 nm amorphous layer.
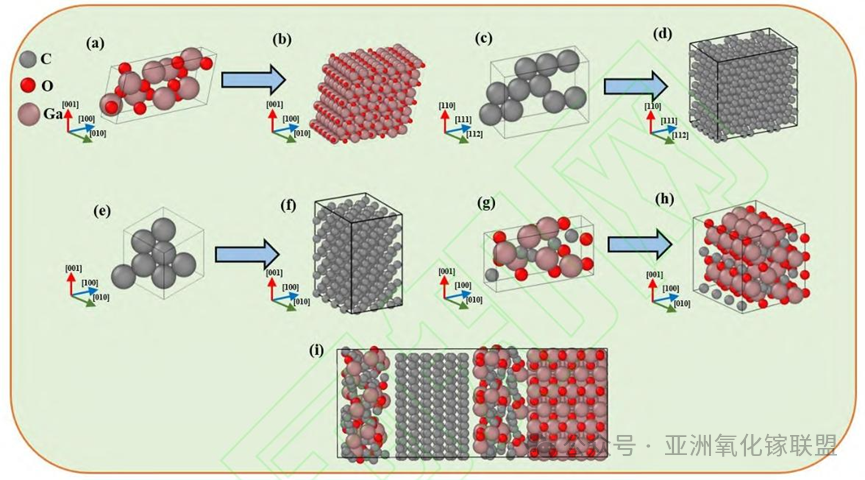
Fig. 3. Atomistic models of the components used in the bonding simulation; (a) β-Ga₂O₃ unit cell; (b) β-Ga₂O₃ supercell; (c) Diamond (111) unit cell; (d) Diamond (111) supercell; (e) Diamond (100) unit cell; (f) Diamond (100) supercell; (g) Amorphous layer unit cell; (h) Amorphous layer supercell; (i) Initial bonding model;
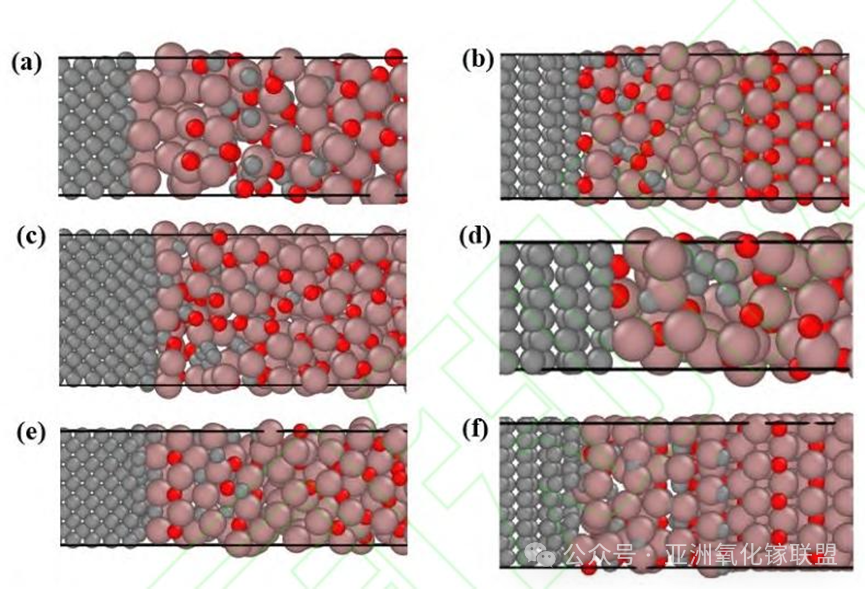
Fig. 4. Bonding interfaces with different crystal orientations; (a) β-Ga2O3 (100)/Diamond(100)interface; (b) β-Ga2O3 (100)/Diamond(111) interface; (c) β-Ga2O3 (010)/Diamond(100) interface;(d) β-Ga2O3 (010)/Diamond(111) interface; (e) β-Ga2O3 (001)/Diamond(100) interface; (f) β-Ga2O3 (001)/Diamond(111) interface;
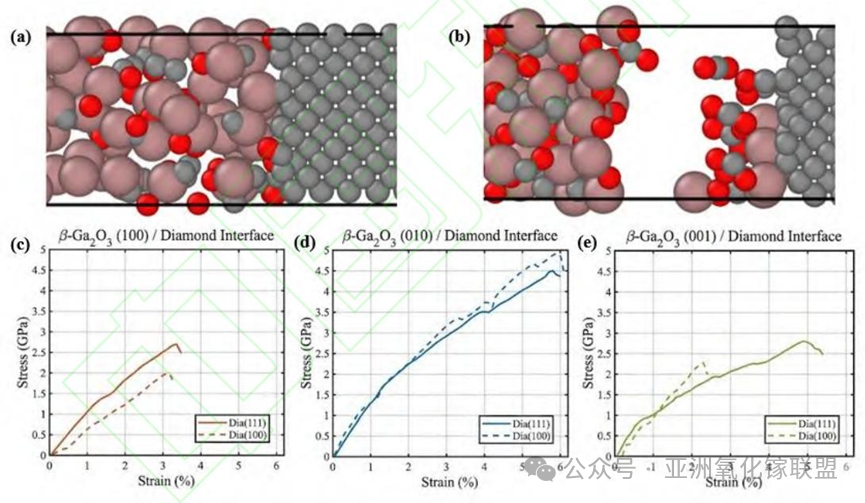
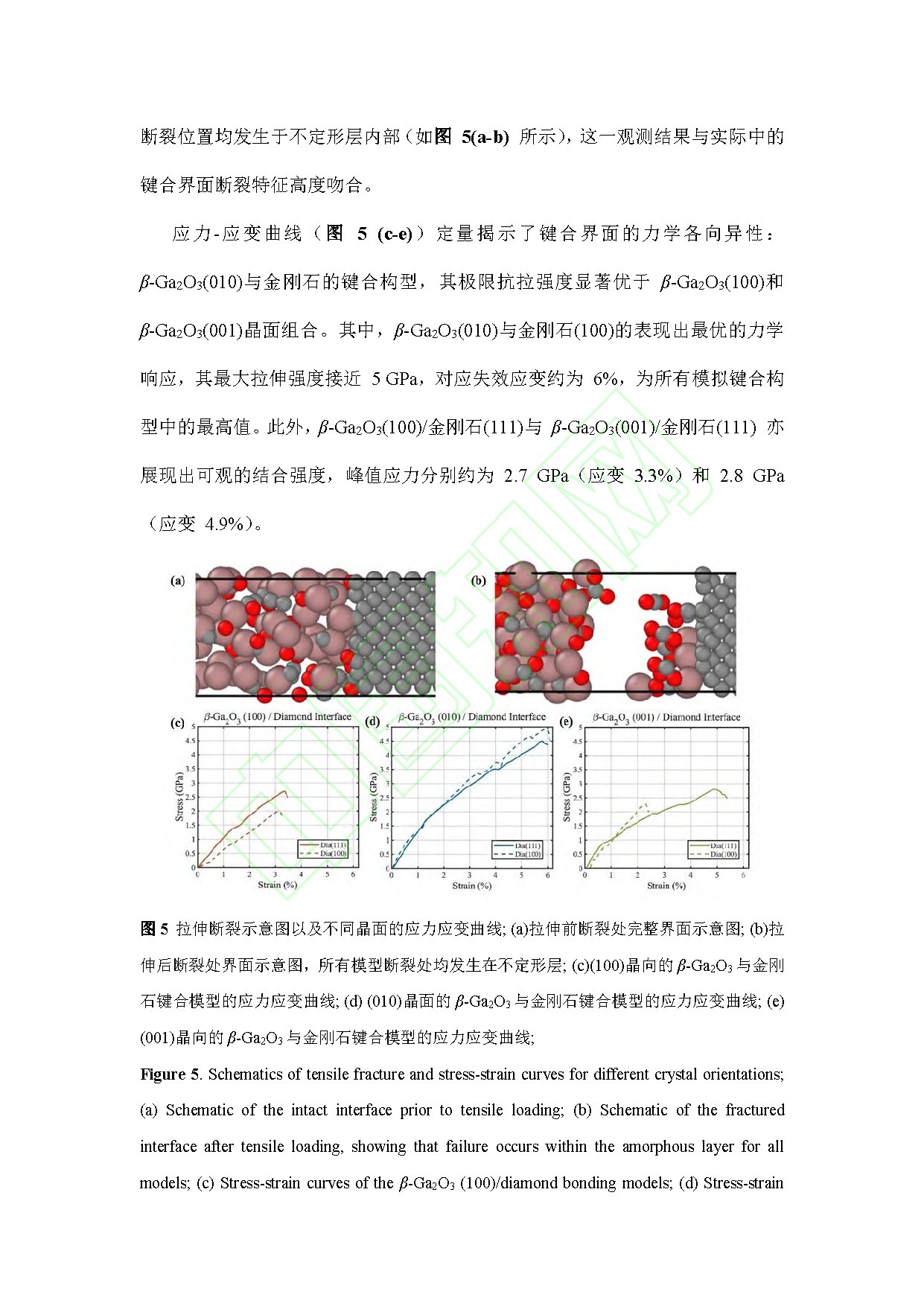
Fig. 5. Schematics of tensile fracture and stress-strain curves for different crystal orientations; (a) Schematic of the intact interface prior to tensile loading; (b) Schematic of the fractured interface after tensile loading, showing that failure occurs within the amorphous layer for all models; (c) Stress-strain curves of the β-Ga₂O₃(100)/diamond bonding models; (d) Stress-strain curves of the β-Ga₂O₃(010)/diamond bonding models; (e) Stress-strain curves of the β-Ga₂O₃(001)/diamond bonding models.
DOI:
10.7498/aps.75.20260116